









vsebina
Cistična fibroza (cistična fibroza)
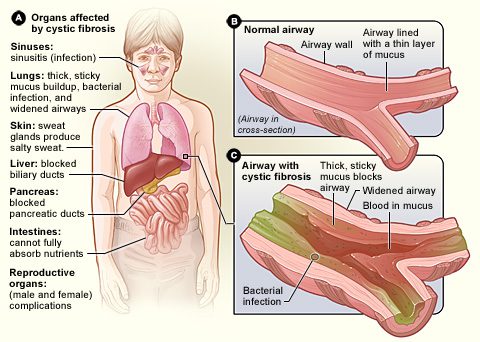
La cistična fibroza, je tukaj genetska bolezen najpogostejši. Glavne manifestacije zadevajo dihala in prebavila, vendar so lahko prizadeti skoraj vsi organi. Simptomi se pogosto pojavijo zgodaj v otroštvu in se razlikujejo po resnosti od osebe do osebe. Ta bolezen povzroča a zgoščevanje sluz, ki jo izločajo sluznice sinusov, bronhijev, črevesja, trebušne slinavke, jeter in reproduktivnega sistema (glejte diagram).
O pljuča so pogosto najbolj prizadeti. The gosti, viskozni izločki ovira bronhije in otežuje dihanje. Poleg tega je sluz, ki se nabira v pljučih, ugodna za rast klic. Ljudje s cistično fibrozo so zato bolj izpostavljeni pogostim in potencialno resnim okužbam dihal.
La cistična fibroza dotika se tudi prebavni sistem. Sluz blokira tanke kanale trebušne slinavke, kar preprečuje, da bi prebavni encimi, ki jih proizvaja trebušna slinavka, vstopili v črevesje in opravljali svojo dejavnost. Ker se hrana le delno prebavi, predvsem maščobe in nekateri vitamini, pride do občutnega pomanjkanja. Lahko povzročijo a zaviranje rasti.
Bolezen ima tudi velike posledice za jetra in reproduktivne organe, kar pogosto povzroči neplodnost pri ženskah in neplodnost pri prizadetih moških.
Zahvaljujoč a zgodnejša diagnoza in boljša oskrba,pričakovana življenjska doba in kakovost življenja prizadetih se je v zadnjih desetletjih še naprej izboljševala, še posebej, ker se začenjajo pojavljati nove terapije, usmerjene v genetsko anomalijo, ki bodo srednjeročno spremenile obravnavo bolnikov. .
Razširjenost
La cistična fibroza ali je genetska bolezen najpogostejši v Franciji s skoraj 6000 prizadetimi ljudmi1.. Vsak četrti novorojenček zboli za to boleznijo. Veliko redkejši je pri temnopoltih (4 v 000) in orientalcih (1 na 13). Prizadene tako moške kot ženske. Najbolj prizadeto je prebivalstvo zahodne Francije.
La cistična fibroza ali je genetska bolezen najpogostejša resna bolezen v Kanadi. Prizadet je eden od 3 novorojenčkov1. Cistična fibroza je nekoliko pogostejša pri Quebec kot v preostali Kanadi: prizadeti so 3 Kanadčani, vključno s 500 Quebeci.
Vzroki
La cistična fibroza je leta 1936 prvič opisal Dr Guido Fanconi, švicarski pediater. Odgovorni gen, imenovan CFTR (za "regulator transmembranske prevodnosti cistične fibroze"), so kanadski raziskovalci identificirali šele leta 1989. Pri bolnih ljudeh to gen is nenormalno (pravimo, da je premeščen). Odgovoren je za sintezo klorovega kanala, ki omogoča uravnavanje hidracije sluzi. V primeru nenormalnosti gena CFTR se sluz izdelek je predebel in ne odteka normalno. Ugotovljena je bila več kot 1 različna mutacija v genu CFTR, ki je vključena v cistično fibrozo2, 3,4. Razdeljeni so v 6 razredov glede na vrsto disfunkcije2Od teh številnih mutacij je najpogostejša mutacija Delta F508, ki jo najdemo pri 81 % prizadetih ljudi v Franciji.
Cistična fibroza ni nalezljiva bolezen. Ljudje, ki imajo v lasti patogene mutacije gena CFTR prej ali slej razvijejo bolezen, vendar v različnih stopnjah resnosti.
Diagnostično
Običajno se cistična fibroza diagnosticira že v prvem letu življenja, ker respiratorni simptomi pojavijo zelo zgodaj. V 90% primerov se bolezen odkrije pred 10. letom starosti.
Za potrditev diagnoze zdravnik opravi a test potenja (ali test potenja). Dejansko je znoja ljudi s cistično fibrozo veliko več koncentriran v soli (2 do 5-krat več kot običajno). The genetski testi omogočajo natančno identifikacijo nepravilnosti v genu CFTR. Bistveni so za razmislek o ciljnih terapijah.
V Franciji so od leta 2002 sistematično pregledovali cistično fibrozo pri vseh novorojenčkih5. Dokazano je, da zgodnje presejanje izboljša kakovost življenja in pričakovano življenjsko dobo prizadetih otrok. Novorojenčki se vzorčijo pri 3 dneh življenja po soglasju staršev, pred odpustom. materinstvo. Test ne daje dokončne diagnoze, ki pa bo potrjena ali razveljavljena s posebnimi dodatnimi preiskavami (test potenja, genetska študija).
V Quebecu ni sistematično presejanje te bolezni. Vendar pa Kanadska fundacija za cistično fibrozo, ki jo podpira več zdravnikov, že nekaj let poziva k izvajanju presejanja novorojenčkov. Dokazano je, da zgodnje odkrivanje izboljša kakovost življenja in pričakovano življenjsko dobo prizadetih otrok.
Pričakovana življenjska doba
V 1960s,pričakovana življenjska doba otrok s cistično fibrozo ni bil starejši od 5 let. Po zadnjih statističnih podatkih je povprečna starost preživetja 47 let1. okužbe dihal ostajajo najpogostejši vzrok smrti.
Pogosti zapleti
Cistična fibroza je bolezen, ki postopoma poškoduje pljuča, trebušno slinavko in jetra. the zdravniški nadzor Vendar pa pomaga zmanjšati resnost in pogostost zapletov.
O respiratorni zapleti so najpogostejši, vključno z razširitvijo bronhijev, ki povzročajo bronhitis, pljučnico s ponovitvami. Pojavijo se obdobja poslabšanja respiratornih simptomov, ko so bolniki zelo »zamašeni«, so bolj zadihani, hujšajo, pogosto zaradi okužbe. Poškodbe dihal so lahko življenjsko nevarne.
Kar zadeva prebavni sistem, obstrukcija žolčevodov, ki omogočajo pretok žolča v prebavni trakt, lahko povzroči cirozo jeter. Obstrukcija in progresivna skleroza trebušna slinavka, lahko povzroči malabsorpcijo hranil in razvoj sladkorne bolezni. Te motnje pogosto vodijo do prehranske pomanjkljivosti huda in kronična driska. Na splošno lahko pomanjkljivosti odpravimo s posebno dieto. Nasprotno pa se lahko pojavi tudi močno zaprtje ali celo črevesna obstrukcija.
Običajno se puberteta pri dečkih in deklicah s cistično fibrozo pojavi pozneje. Končno, plodnost je zmanjšala, zlasti pri moških, ki so skoraj vsi (95 %) sterilni zaradi obstrukcije semenovoda. Ti kanali prenašajo spermo iz mod v semenske mehurčke. Pri ženskah povečana viskoznost vaginalne sluzi upočasni gibanje sperme. Bolezen lahko vpliva tudi na pravilnost in pogostost ovulacije. Plodnost upada, vendar je nosečnost še vedno povsem možna.